Publications
publications by categories in reversed chronological order.
2025
-
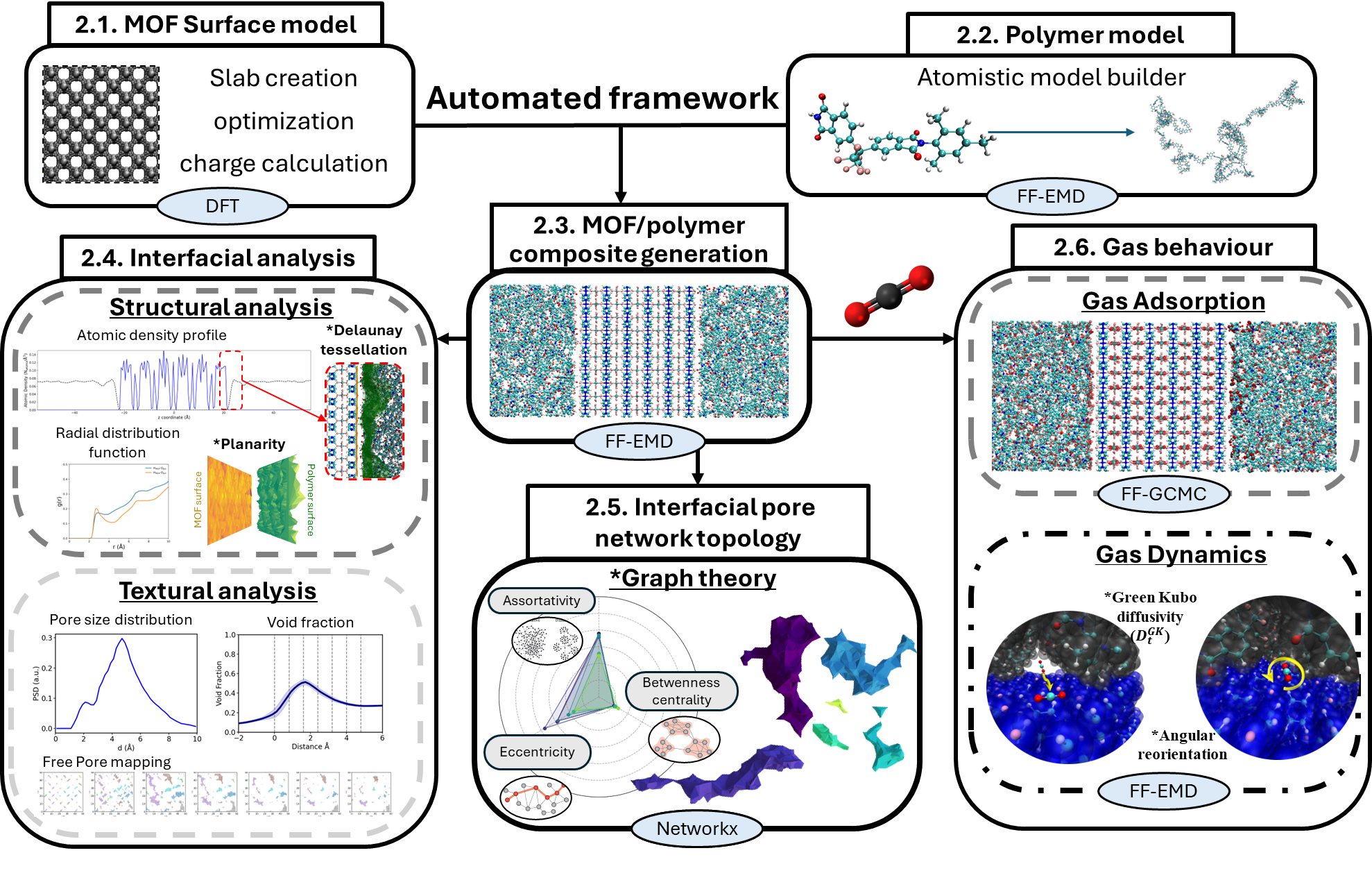 MOF surface morphology governs interfacial pore architecture and CO₂ dynamics in mixed matrix membranesAlejandro Diaz-Marquez, Supriyo Naskar, Dong Fan, and 2 more authorsChemical Science, Oct 2025
MOF surface morphology governs interfacial pore architecture and CO₂ dynamics in mixed matrix membranesAlejandro Diaz-Marquez, Supriyo Naskar, Dong Fan, and 2 more authorsChemical Science, Oct 2025"Mixed matrix membranes (MMMs), which embed metal–organic frameworks (MOFs) within polymers, offer a promising platform for next-generation, energy-efficient separations. However, the nano-structuring of the MOF/polymer interface and its influence on the MMM performance remains poorly understood. Here, we uncover two fundamental design principles that bridge this gap, enabled by an automated graph theory-enhanced molecular simulation platform. First, we demonstrate that MOF surface morphology, specifically its planarity and roughness, plays a decisive role in shaping the topology of the interfacial pore network, including its dimensionality, connectivity, and spatial organization. Second, we show that this pore topology critically governs interfacial CO₂ dynamics: highly interconnected and continuous networks facilitate efficient translational and rotational motion, whereas fragmented architectures severely limit molecular mobility. Beyond providing a deep molecular-level understanding, this work introduces a new design paradigm: deliberate tuning of MOF surface morphology emerges as a powerful strategy to control interfacial nanostructure and optimize gas dynamics. Together, these findings open an unexplored pathway for the rational design of high-performance MMMs for advancing energy-efficient separation technologies."
- ACS
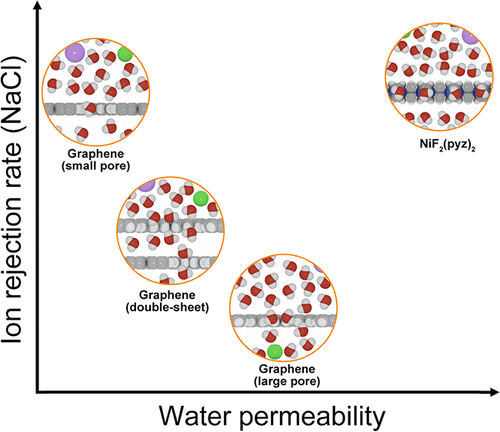 Intrinsic Pore Architecture of a Two-Dimensional MOF: A Blueprint for Overcoming the Permeability-Selectivity Trade-Off in DesalinationDong Fan, Ke Zheng, Supriyo Naskar, and 1 more authorChemistry of Materials, Nov 2025
Intrinsic Pore Architecture of a Two-Dimensional MOF: A Blueprint for Overcoming the Permeability-Selectivity Trade-Off in DesalinationDong Fan, Ke Zheng, Supriyo Naskar, and 1 more authorChemistry of Materials, Nov 2025Water scarcity remains one of the most pressing global challenges, driving the urgent need for next-generation desalination technologies that can overcome the long-standing trade-off between water permeability and ion selectivity. Here, we introduce a molecular design blueprint based on a two-dimensional metal−organic framework (2D-MOF) monolayer, NiF2(pyz)2, featuring intrinsically aligned ∼4 Å pores and high structural regularity. Using systematic all-atom molecular dynamics simulations, we demonstrate that this monolayer achieves ultrahigh water permeability while maintaining 100% Na+/Cl− rejection across a wide range of applied pressures. We attribute this exceptional performance to a synergistic combination of low interfacial water density, reduced free energy barriers for water transport, and a high pore area-to-surface area ratio, captured by a proposed dimensionless descriptor. Compared to conventional 2D membranes with artificial nanopores, which often suffer from fabrication complexity, pore size variability, and mechanical fragility, the NiF2(pyz)2 monolayer offers a robust alternative with precisely defined nanochannels combined with mechanical and hydrothermal stability. This work pushes the boundaries of the structure−function paradigm in membrane science by establishing that atomically engineered intrinsic porosity, rather than postfabricated artificial channels, is the key to achieving fast and selective water transport. Our findings lay the groundwork for the rational design of high-efficiency desalination membranes and suggest possible approaches for using MOFs in sustainable water purification technologies.


2024
- Nature
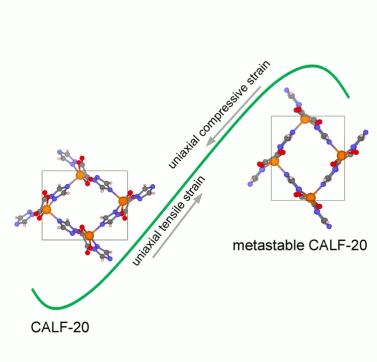 Unconventional mechanical and thermal behaviors of MOF CALF-20Dong Fan, Supriyo Naskar, and Guillaume MaurinNature Communications, Jan 2024
Unconventional mechanical and thermal behaviors of MOF CALF-20Dong Fan, Supriyo Naskar, and Guillaume MaurinNature Communications, Jan 2024CALF-20 was recently identified as a novel benchmark sorbent for CO2 capture at the industrial scale, however comprehensive atomistic insight into its mechanical/thermal properties under working conditions is still lacking. In this study, we developed a general-purpose machine-learned potential (MLP) for the CALF-20 MOF framework that predicts the thermodynamic and mechanical properties of the structure at finite temperatures within first-principles accuracy. Interestingly, CALF-20 was demonstrated to exhibit both negative area compression and negative thermal expansion. Most strikingly, upon application of the tensile strain along the [001] direction, CALF-20 was shown to display a distinct two-step elastic deformation behavior, unlike typical MOFs that undergo plastic deformation after elasticity. Furthermore, this MOF was shown to exhibit a spectacular fracture strain of up to 27% along the [001] direction at room temperature comparable to that of MOF glasses. These abnormal thermal and mechanical properties make CALF-20 as attractive material for flexible and stretchable electronics and sensors.
- ACS
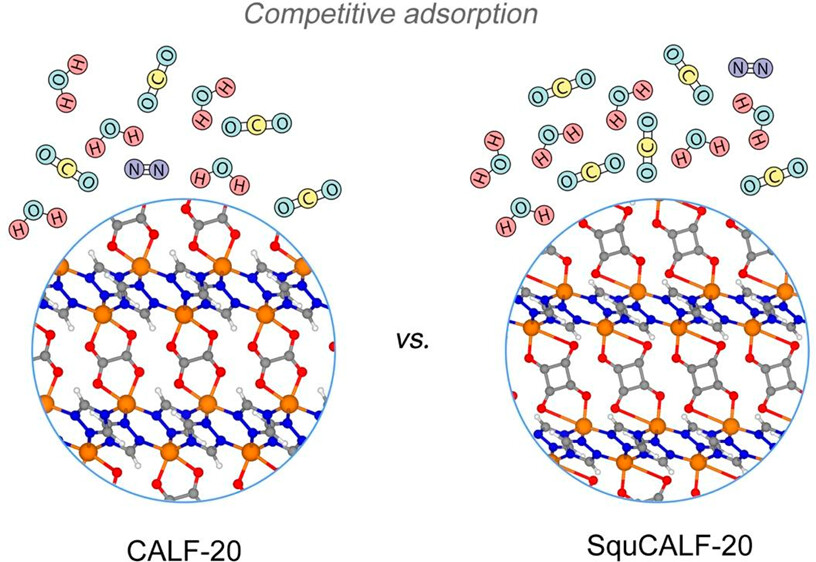 Engineering of an Isoreticular Series of CALF-20 Metal–Organic Frameworks for CO2 CaptureKaruppasamy Gopalsamy, Dong Fan, Supriyo Naskar, and 2 more authorsACS Applied Engineering Materials, Jan 2024
Engineering of an Isoreticular Series of CALF-20 Metal–Organic Frameworks for CO2 CaptureKaruppasamy Gopalsamy, Dong Fan, Supriyo Naskar, and 2 more authorsACS Applied Engineering Materials, Jan 2024A series of linker-substituted ultramicroporous CALF-20 metal–organic frameworks (MOFs) were built in silico, and their CO_2 capture performances over N2 in flue gas conditions were systematically computationally explored. Among the various linker substitutions explored, squarate-linker-incorporated CALF-20 (SquCALF-20) was demonstrated to show a larger CO2 uptake at 0.15 bar (3.6 mmol/g) and higher CO2/N2 selectivity (500) in dry conditions compared to pristine CALF-20. Interestingly, this MOF was shown to maintain a high level of CO2 capture performance even in the presence of humidity, although it starts to adsorb H2O at lower relative humidity compared to CALF-20. Because squaric acid is a semiconductor industry feedstock and the few-already published squarate-based MOFs are chemically robust, this engineered SquCALF-20 offers a promising avenue for cost-effective CO2 capture via physisorption, with potential applications in addressing environmental concerns associated with CO2 emissions.
- Wiley
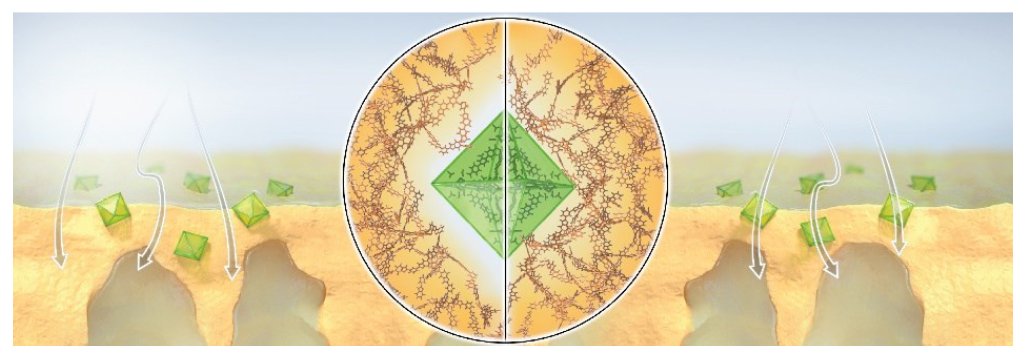 Design of Mixed-Matrix MOF Membranes with Asymmetric Filler Density and Intrinsic MOF/Polymer Compatibility for Enhanced Molecular SievingRifan Hardian, Jiangtao Jia, Alejandro Diaz-Marquez, and 6 more authorsAdvanced Materials, Jan 2024
Design of Mixed-Matrix MOF Membranes with Asymmetric Filler Density and Intrinsic MOF/Polymer Compatibility for Enhanced Molecular SievingRifan Hardian, Jiangtao Jia, Alejandro Diaz-Marquez, and 6 more authorsAdvanced Materials, Jan 2024The separation of high-value-added chemicals from organic solvents is of prime importance for many industries but it is associated with a large energy consumption penalty. Suitably, membrane-based nanofiltration offers potential for a more energy-efficient separation than the conventional energy-intensive thermal processes. Conceivably, mixed-matrix membranes (MMMs), encompassing metal-organic frameworks (MOFs) as fillers, are poised to promote selective separation via molecular sieving, synergistically combining polymers flexibility and fine-tuned porosity of MOFs. Nevertheless, conventional direct mixing of MOFs with polymer solutions results in underutilization of the MOF fillers owing to their uniform cross-sectional distribution. Therefore, in this work, we propose a multizoning technique for the formation of MMMs with an asymmetric-filler density at the macroscale, in which the MOF fillers are distributed only on the surface of the membrane, and a seamless interface at the nanoscale. Our design strategy demonstrates five times higher MOF surface coverage, which results in a solvent permeance five times higher than that of conventional MMMs while maintaining high selectivity. Practically, MOFs are paired with polymers of similar chemical nature in order to enhance their adhesion without the need for additional surface modification. Our approach offers permanently accessible MOF porosity, which translates to effective molecular sieving, as exemplified by the polybenzimidazole and Zr-BI-fcu-MOF system. Our findings pave the way for the development of composite materials with a seamless interface.



2023
- RSC
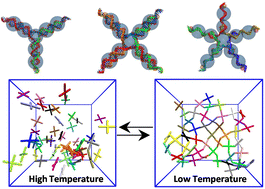 Mechanistic insight into the structure, thermodynamics and dynamics of equilibrium gels of multi-armed DNA nanostarsSupriyo Naskar, Dhiraj Bhatia, Shiang-Tai Lin, and 1 more authorPhysical Chemistry Chemical Physics, Feb 2023
Mechanistic insight into the structure, thermodynamics and dynamics of equilibrium gels of multi-armed DNA nanostarsSupriyo Naskar, Dhiraj Bhatia, Shiang-Tai Lin, and 1 more authorPhysical Chemistry Chemical Physics, Feb 2023The unique sequence specificity rule of DNA makes it an ideal molecular building block for constructing periodic arrays and devices with nanoscale accuracy and precision. Here, we present the self-assembly of DNA nanostars having three, four and five arms into a gel phase using a simplistic coarse-grained bead-spring model developed by Z. Xing, C. Ness, D. Frenkel and E. Eiser (Macromolecules, 2019, 52, 504–512). Our simulations show that the DNA nanostars form a thermodynamically stable fully bonded gel phase from an unstructured liquid phase with the lowering of temperature. We characterize the phase transition by calculating several structural features such as the radial distribution function and structure factor. The thermodynamics of gelation is quantified by the potential energy and translational pair-entropy of the system. The phase transition from an arrested gel phase to an unstructured liquid phase has been modelled using a two-state theoretical model. We find that this transition is enthalpy driven, and loss of configuration and translational entropy is counterpoised by enthalpic interaction of the DNA sticky-ends, which gives rise to a gel phase at low temperature. The absolute rotational and translational entropy of the systems, measured using a two-phase thermodynamic model, also substantiates the gel transition. The slowing down of the dynamics upon approaching the transition temperature from a high temperature demonstrates the phase transition to a gel phase. A detailed numerical simulation study of the morphology, dynamics and thermodynamics of DNA gelation can provide guidance for future experiments, is easily extensible to other polymeric systems, and is expected to help in understanding the physics of self-assembly.
- RSC
 Microscopic insight into the shaping of MOFs and its impact on CO 2 capture performanceSupriyo Naskar, Dong Fan, Aziz Ghoufi, and 1 more authorChemical Science, Sep 2023
Microscopic insight into the shaping of MOFs and its impact on CO 2 capture performanceSupriyo Naskar, Dong Fan, Aziz Ghoufi, and 1 more authorChemical Science, Sep 2023The traditional synthesis method produces microcrystalline powdered MOFs, which prevents direct implementation in real-world applications which demand strict control of shape, morphology and physical properties. Therefore, shaping of MOFs via the use of binders is of paramount interest for their practical use in gas adsorption/separation, catalysis, sensors, etc. However, so far, the binders have been mostly selected by trial-and-error without anticipating the adhesion between the MOF and binder components to ensure the processability of homogeneous and mechanically stable shaped MOFs and the impact of the shaping on the intrinsic properties of the MOFs has been overlooked. Herein, we deliver a first systematic multiscale computational exploration of MOF/binder composites by selecting CALF-20, a prototypical MOF for real application in the field of CO2 capture, and a series of binders that cover a rather broad spectrum of properties in terms of rigidity/flexibility, porosity, and chemical functionality. The adhesion between the two components and hence the effectiveness of the shaping as well as the impact of the overall porosity of the CALF-20/binder on the CO2/N2 selectivity, CO2 sorption capacity and kinetics was analyzed. Shaping of CALF-20 by carboxymethyl cellulose was predicted to enable a fair compromise between excellent adhesion between the two components, whilst maintaining high CO2/N2 selectivity, large CO2 uptake and CO2 transport as fast as in the CALF-20. This multiscale computational tool paves the way towards the selection of an appropriate binder to achieve an optimum shaping of a given MOF in terms of processability whilst maintaining its high level of performance.


2022
- RSC
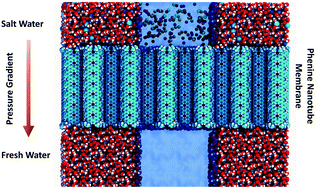 Ultra-high permeable phenine nanotube membranes for water desalinationSupriyo Naskar, Anil Kumar Sahoo*, Mohd Moid*, and 1 more authorPhysical Chemistry Chemical Physics, Apr 2022
Ultra-high permeable phenine nanotube membranes for water desalinationSupriyo Naskar, Anil Kumar Sahoo*, Mohd Moid*, and 1 more authorPhysical Chemistry Chemical Physics, Apr 2022Nanopore desalination technology hinges on high water-permeable membranes which, at the same time, block ions efficiently. In this study, we consider a recently synthesized [Science 363, 151–155 (2019)] phenine nanotube (PNT) for water desalination applications. Using both equilibrium and non-equilibrium molecular dynamics simulations, we show that the PNT membrane completely rejects salts, but permeates water at a rate which is an order-of-magnitude higher than that of all the membranes used for water filtration. We provide the microscopic mechanisms of salt rejection and fast water-transport by calculating the free-energy landscapes and electrostatic potential profiles. A collective diffusion model accurately predicts the water permeability obtained from the simulations over a wide range of pressure gradients. We propose a method to calculate the osmotic water permeability from the equilibrium simulation data and find that it is very high for the PNT membrane. These remarkable properties of PNT can be applied in various nanofluidic applications, such as ion-selective channels, ionic transistors, sensing, molecular sieving, and blue energy harvesting.
- ACS
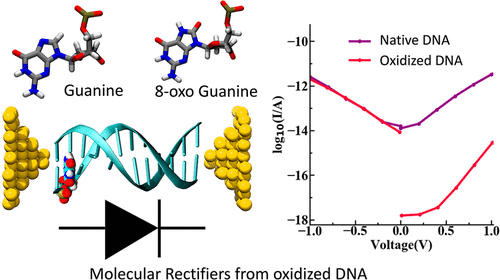 Molecular rectifiers with very high rectification ratio enabled by oxidative damage in double-stranded DNAAbhishek Aggarwal, Supriyo Naskar, and Prabal K. MaitiThe Journal of Physical Chemistry B, Mar 2022
Molecular rectifiers with very high rectification ratio enabled by oxidative damage in double-stranded DNAAbhishek Aggarwal, Supriyo Naskar, and Prabal K. MaitiThe Journal of Physical Chemistry B, Mar 2022In this work, we report a novel strategy to construct molecular diodes with a record tunable rectification ratio of as high as 10^6 using oxidatively damaged DNA molecules. Being exposed to several endogenous and exogenous events, DNA suffers constant oxidative damages leading to oxidation of guanine to 8-Oxoguanine (8oxoG). Here, we study the charge migration properties of native and oxidatively damaged DNA using a multiscale multiconfigurational methodology comprising of molecular dynamics, density functional theory and kinetic Monte Carlo simulations. We perform a comprehensive study to understand the effect of different concentrations and locations of 8oxoG in a dsDNA sequence on its CT properties and find tunable rectifier properties having potential applications in molecular electronics such as molecular switches and molecular rectifiers. We also discover the negative differential resistance properties of fully oxidized Drew-Dickerson sequence. The presence of 8oxoG guanine leads to the trapping of charge, thus operates as a charge sink, which reveals how oxidized guanine saves the rest of the genome from further oxidative damage.
- RSC
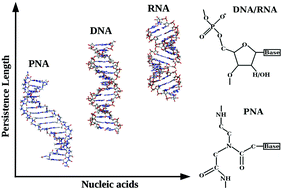 Nanoscale Structure and Mechanics of Peptide Nucleic AcidsKhadka Bahadur Chhetri, Akshara Sharma, Supriyo Naskar, and 1 more authorNanoscale, Apr 2022
Nanoscale Structure and Mechanics of Peptide Nucleic AcidsKhadka Bahadur Chhetri, Akshara Sharma, Supriyo Naskar, and 1 more authorNanoscale, Apr 2022Peptide nucleic acids (PNAs) are charge-neutral polyamide oligomers having extremely favorable thermal stability and high affinity to cell membranes when coupled with cationic cell-penetrating peptides (CPPs), as well as the encouraging antisense and antigene activity in cell-free systems. The study of the mechanical properties of short PNA molecules is rare both in experiments and theoretical calculations. Here, we studied the microscopic structures and elastic properties; namely, persistence length, stretch modulus, twist–stretch coupling, and structural crookedness of double-stranded PNA (dsPNA) and their hybrid derivatives using all-atom MD simulation and compared them with those of double-stranded DNA (dsDNA) and double-stranded RNA (dsRNA). The stretch modulus of the dsPNA is found to be ∼160 pN, an order of magnitude lower than that of dsDNA and smaller than dsRNA, respectively. Similarly, the persistence length of dsPNA is found to be ∼35 nm, significantly smaller than those of dsDNA and dsRNA. The PNA–DNA and PNA–RNA hybrid duplexes have elastic properties lying between that of dsPNA and dsDNA/dsRNA. We argue that the neutral backbones of the PNA make it less stiff than dsDNA and dsRNA molecules. Measurement of structural crookedness and principal component analysis additionally support the bending flexibility of dsPNA. Detailed analysis of the helical-rise coupled to helical-twist indicates that the PNA–DNA hybrid over-winds like dsDNA, while PNA–PNA and PNA–RNA unwind like dsRNA upon stretching. Because of the highly flexible nature of PNA, it can bind other biomolecules by adopting a wide range of conformations and is believed to be crucial for future nanobiotechnology research studies.
- Springer
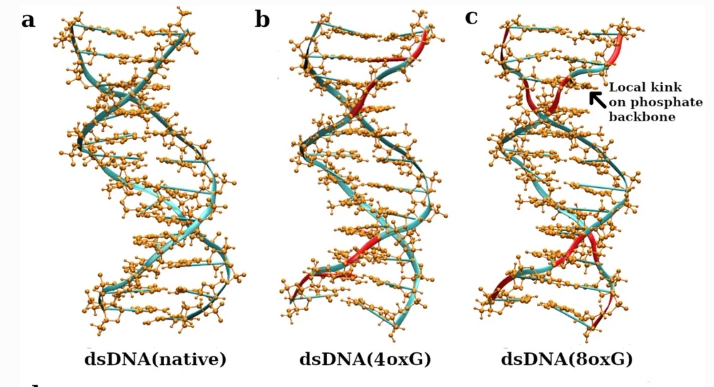 Probing the microscopic structure and flexibility of oxidized DNA by molecular simulationsKhadka B Chhetri, Supriyo Naskar, and Prabal K. MaitiIndian Journal of Physics, Mar 2022
Probing the microscopic structure and flexibility of oxidized DNA by molecular simulationsKhadka B Chhetri, Supriyo Naskar, and Prabal K. MaitiIndian Journal of Physics, Mar 2022The oxidative damage of DNA is a compelling issue in molecular biophysics as it plays a vital role in the epigenetic control of gene expression and is believed to be associated with mutagenesis, carcinogenesis and aging. To understand the microscopic structural changes in physical properties of DNA and the resulting influence on its function due to oxidative damage of its nucleotide bases, we have conducted all-atom molecular dynamic simulations of double-stranded DNA (dsDNA) with its guanine bases being oxidized. The guanine bases are more prone to oxidative damage due to the lowest value of redox potential among all nucleobases. We have analyzed the local as well as global mechanical properties of native and oxidized dsDNA and explained those results by microscopic structural parameters and thermodynamic calculations. Our results show that the oxidative damage of dsDNA does not deform the Watson-Crick geometry; instead, the oxidized DNA structures are found to be better stabilized through electrostatic interactions. Moreover, oxidative damage changes the mechanical, helical and groove parameters of dsDNA. The persistence length, stretch modulus and torsional stiffness are found to be 48.87 nm, 1239.26 pN and 477.30 pN.nm2, respectively, for native dsDNA and these values are 61.31 nm, 659.91 pN and 407.79 pN.nm2, respectively, when all the guanine bases of the dsDNA are oxidized. Compared to the global mechanical properties, the changes in helical and groove properties are found to be more prominent, concentrated locally at the oxidation sites and causing the transition of the backbone conformations from BI to BII at the regions of oxidative damage.
- RSC
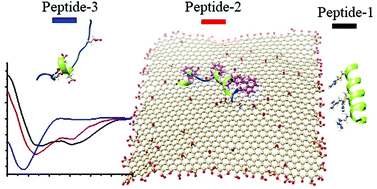 Graphene Oxide as a Dual Template for Induced Helicity of PeptidesSubrata Pandit, Nikhil Maroli, Supriyo Naskar, and 3 more authorsNanoscale, May 2022
Graphene Oxide as a Dual Template for Induced Helicity of PeptidesSubrata Pandit, Nikhil Maroli, Supriyo Naskar, and 3 more authorsNanoscale, May 2022An artificial template mediated fabrication of secondary structure within peptides always attracts great interest in biological system due to its several biomimetic interactions. In all earlier studies, an uniform template containing molecules/nanomaterials were used to target only one type of peptide at a time which extensively limits the diversity in the generation of artificial protein surface/receptors. This limitation can be overcome by the incorporation of more than one binding template (heterogenity) in a single system, for example Janus nanomaterials, which is challenging and difficult to synthesize. In this context, graphene oxide (GO) was considered as an artificial receptor (template). It contains two distinctive binding zones i.e., surface and edge, which can induce the secondary structure of peptides based on complimentary interactions. To establish our concept, we have implemented a hybrid sequence i.e., i, i+4, i+7 and i+11 pattern peptides which defines a more linear surface, suitable for recognition by the two-dimensional GO. Depending on the amino acid residue at the specific locations, we observed substantial enhancement of peptide helicity either at the surface or edges of GO from the random coil. However, non-interacting peptide remains as random coil. We have established this by circular dichroism study at various conditions, as well as atomic force microscopy and optical imaging study. Furthermore, we have also established our observation using molecular dynamics (MD) simulations. This study reveals that synthesized GO-peptides composite with different secondary structures and recognition residues can mimic biological systems.





2021
- RSC
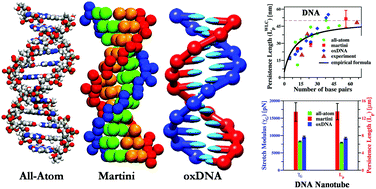 Mechanical properties of DNA and DNA nanostructures: comparison of atomistic, Martini and oxDNASupriyo Naskar, and Prabal K. MaitiJournal of Materials Chemistry B, May 2021
Mechanical properties of DNA and DNA nanostructures: comparison of atomistic, Martini and oxDNASupriyo Naskar, and Prabal K. MaitiJournal of Materials Chemistry B, May 2021The flexibility and stiffness of small DNA molecules play a fundamental role ranging from several biophysical processes to nano-technological applications. Here, we estimate the mechanical properties of short double-stranded DNA (dsDNA) with lengths ranging from 12 base-pairs (bp) to 56 bp, paranemic crossover (PX) DNA and hexagonal DNA nanotubes (DNTs) using two widely used coarse-grained models – Martini and oxDNA. To calculate the persistence length (Lp) and the stretch modulus (γ) of the dsDNA, we incorporate the worm-like chain and elastic rod model, while for the DNTs, we implement our previously developed theoretical framework. We compare and contrast all of the results with previously reported all-atom molecular dynamics (MD) simulations and experimental results. The mechanical properties of dsDNA (Lp ∼ 50 nm, γ ∼ 800–1500 pN), PX DNA (γ ∼ 1600–2000 pN) and DNTs (Lp ∼ 1–10 μm, γ ∼ 6000–8000 pN) estimated using the Martini soft elastic network and oxDNA are in very good agreement with the all-atom MD and experimental values, while the stiff elastic network Martini reproduces values of Lp and γ which are an order of magnitude higher. The high flexibility of small dsDNA is also depicted in our calculations. However, Martini models proved inadequate to capture the salt concentration effects on the mechanical properties with increasing salt molarity. oxDNA captures the salt concentration effect on the small dsDNA mechanics. But it is found to be ineffective for reproducing the salt-dependent mechanical properties of DNTs. Also, unlike Martini, the time evolved PX DNA and DNT structures from the oxDNA models are comparable to the all-atom MD simulated structures. Our findings provide a route to study the mechanical properties of DNA and DNA based nanostructures with increased time and length scales and has a remarkable implication in the context of DNA nanotechnology.
- ACS
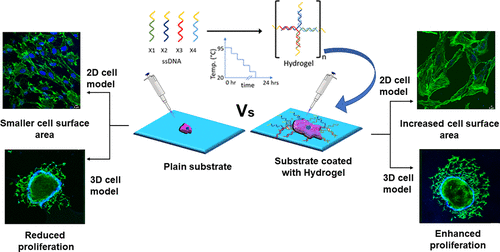 Designer DNA hydrogels to stimulate 3d cell invasion by enhanced receptor expression and membrane endocytosisShanka Walia, Vinod Morya, Ankit Gangrade, and 6 more authorsACS Biomaterials Science & Engineering, Dec 2021
Designer DNA hydrogels to stimulate 3d cell invasion by enhanced receptor expression and membrane endocytosisShanka Walia, Vinod Morya, Ankit Gangrade, and 6 more authorsACS Biomaterials Science & Engineering, Dec 2021DNA has emerged as one of the smartest biopolymers to bridge the gap between chemical science and biology to design scaffolds like hydrogels by physical entanglement or chemical bonding with remarkable properties. We present here a completely new application of DNA based hydrogels in terms of their capacity to stimulate membrane endocytosis, leading to enhanced cell spreading and invasion for cells in ex-vivo 3D spheroids models. Multiscale simulation studies along with DLS data showed that the hydrogel formation was enhanced at lower temperature and it converts to liquid with increase in temperature. DNA hydrogels induced cell spreading as observed by increase in cellular area by almost two-folds followed by increase in receptor expression, endocytosis and 3D invasion potential of migrating cells. Our first results lay the foundation for upcoming diverse applications of hydrogels to probe and program various cellular and physiological processes that can have lasting applications in stem cells programming and regenerative therapeutics.
- RSC
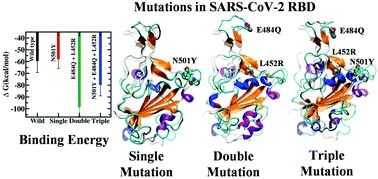 Mechanistic Insights into the Effects of Key Mutations on SARS-CoV-2 RBD-ACE2 BindingAbhishek Aggarwal*, Supriyo Naskar*, Nikhil Maroli*, and 3 more authorsPhysical Chemistry Chemical Physics, Nov 2021
Mechanistic Insights into the Effects of Key Mutations on SARS-CoV-2 RBD-ACE2 BindingAbhishek Aggarwal*, Supriyo Naskar*, Nikhil Maroli*, and 3 more authorsPhysical Chemistry Chemical Physics, Nov 2021Some recent SARS-CoV-2 variants appear to have increased transmissibility compared to the original strain. An underlying mechanism could be the improved ability of the variants to bind receptors on the target cells and infect them. In this study, we provide atomic-level insights into the binding of the receptor binding domain (RBD) of the wild-type SARS-CoV-2 spike protein and its single (N501Y), double (E484Q, L452R) and triple (N501Y, E484Q, L452R) mutated variants to the human ACE2 receptor. Using extensive all-atom molecular dynamics simulations and advanced free energy calculations, we estimate the associated binding affinities and binding hotspots. We observe significant secondary structural changes in the RBD of the mutants, which lead to different binding affinities. We find higher binding affinities for the double (E484Q, L452R) and triple (N501Y, E484Q, L452R) mutated variants than for the wild type and the N501Y variant, which could contribute to the higher transmissibility of recent variants containing these mutations.
- IISc
 Multiscale Simulation of Nucleic Acid NanostructuresSupriyo NaskarPhD Thesis Indian Institute of Science, Nov 2021
Multiscale Simulation of Nucleic Acid NanostructuresSupriyo NaskarPhD Thesis Indian Institute of Science, Nov 2021Nucleic acids, namely DNA and RNA, are arguably the most studied biological molecules. Utilizing the key properties of nucleic acids, such as their persistence lengths and Watson-Crick base-pairing complementarity, nanometer-scale devices and other functional materials can be created. Numerous experimental studies have been performed on these nanostructures both in vitro as well as in vivo. However, in many cases, it is hard to decipher their in-situ structures, interpret molecular level understanding of their self-assembly and understand many experimental observations, particularly those that originate from structural changes at the single-molecule level. In this thesis, using multiscale molecular dynamics simulations, we have studied the structure, mechanics, and thermodynamics of various self-assembled and artificial nucleic acid nanostructures. In particular, we have developed a de novo computational framework to model atomistic as well as coarse-grained nucleic acid nanotubes. Using various theoretical models, we have estimated their microscopic structure, mechanical properties and found excellent agreement with available values from the experiments. We have also studied a minimal bead-spring coarse-grained model of DNA nanostars which self-assemble into complex polymeric network known as DNA hydrogel at low temperature. The phase transition from an unstructured fluid to a gel-like network structure with the lowering of temperature has been explained by calculations of structural parameters, thermodynamic quantities and dynamics of the systems. Finally, the microscopic origin of liquid crystal ordering of ultra-short nucleic acids that do not satisfy the shape anisotropy criterion of Onsager’s cylindrical rods is studied. Employing advanced free-energy calculation techniques, we observe that ultra-short nucleic acids prefer to stack on top of each other while repelling sideways, leading to the formation of supramolecular columns that undergo LC ordering at high volume fraction. The study presented in this thesis has showcased the ability of multiscale molecular simulation and statistical physics to predict the structural, mechanical, and thermodynamic properties of various self-assembled and engineered DNA nanostructures.




2020
- RSC
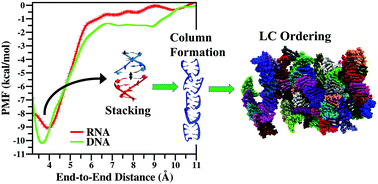 Liquid crystal ordering of nucleic acidsSupriyo Naskar, Suman Saurabh, Yun Hee Jang, and 2 more authorsSoft Matter, Jan 2020
Liquid crystal ordering of nucleic acidsSupriyo Naskar, Suman Saurabh, Yun Hee Jang, and 2 more authorsSoft Matter, Jan 2020Several analytical calculations and computer simulations propose that cylindrical monodispersive rods having an aspect ratio (ratio of length to diameter) greater than 4 can exhibit liquid crystal (LC) ordering. But, recent experiments demonstrated the signature of LC ordering in systems of 4- to 20-base pair (bp) long nucleic acids (NAs) that do not satisfy the shape anisotropy criterion. Mechanisms of end-to-end adhesion and stacking have been proposed to explain this phenomenon. In this study, using all-atom molecular dynamics (MD) simulation, we explicitly verify the end-to-end stacking of double-stranded RNA (dsRNA) and demonstrate the LC ordering at the microscopic level. Using umbrella sampling (US) calculation, we quantify the potential of mean force (PMF) between two dsRNAs for various reaction coordinates (RCs) and compare our results with previously reported PMFs for double-stranded DNA (dsDNA). The PMF profiles demonstrate the anisotropic nature of inter-NA interaction. We find that, like dsDNA, dsRNA also prefers to stack on top of each other while repelling sideways, leading to the formation of supra-molecular-columns that undergo LC ordering at high NA volume fraction. We also demonstrate and quantify the nematic ordering of the RNAs using several hundred nanosecond-long MD simulations that remain almost invariant for different initial configurations and under different external physiological conditions.
- Elsevier
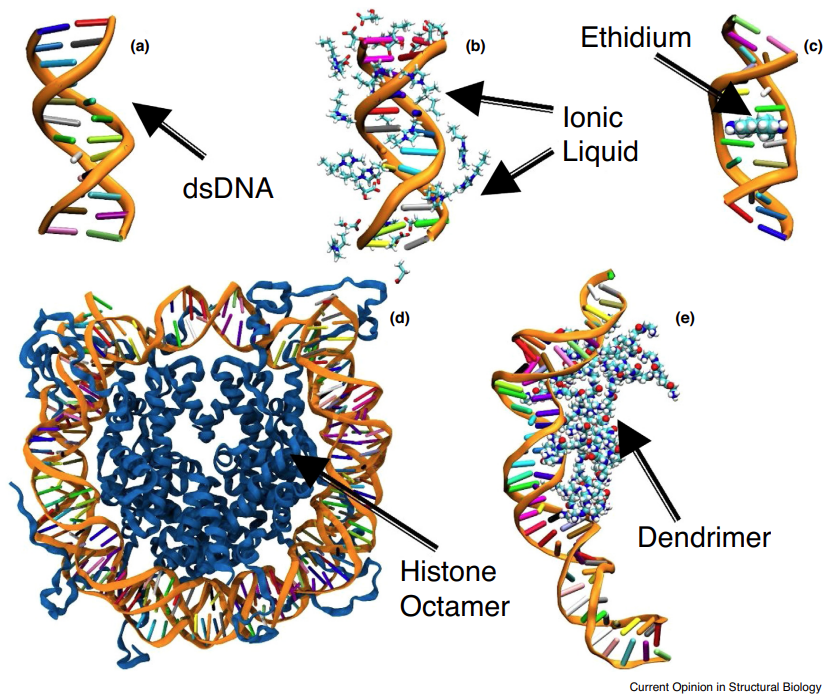 What do we know about DNA mechanics so far?Abhishek Aggarwal*, Supriyo Naskar*, Anil Kumar Sahoo*, and 3 more authorsCurrent opinion in structural biology, Oct 2020
What do we know about DNA mechanics so far?Abhishek Aggarwal*, Supriyo Naskar*, Anil Kumar Sahoo*, and 3 more authorsCurrent opinion in structural biology, Oct 2020The DNA molecule, apart from carrying the genetic information, plays a crucial role in a variety of biological processes and finds applications in drug design, nanotechnology and nanoelectronics. The molecule undergoes significant structural transitions under the influence of forces due to physiological and non-physiological environments. Here, we summarize the insights gained from simulations and single-molecule experiments on the structural transitions and mechanics of DNA under force, as well as its elastic properties, in various environmental conditions, and discuss appealing future directions.


2019
- ACS
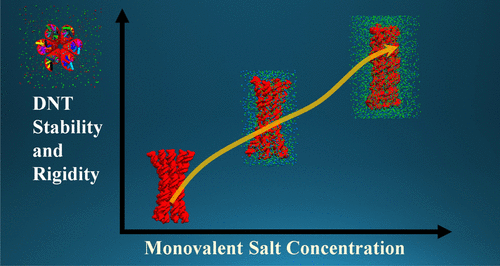 Tuning the Stability of DNA Nanotubes with SaltSupriyo Naskar, Mounika Gosika, Himanshu Joshi, and 1 more authorThe Journal of Physical Chemistry C, Mar 2019
Tuning the Stability of DNA Nanotubes with SaltSupriyo Naskar, Mounika Gosika, Himanshu Joshi, and 1 more authorThe Journal of Physical Chemistry C, Mar 2019We report the enhancement of the structural stability of a DNA nanotube (DNT) by changing the salt concentrations for three different salt species, namely, NaCl, KCl, and MgCl2. Using fully atomistic molecular dynamics simulations, we find that, with the gradual increment in the NaCl salt concentration, the DNT becomes compact and rigid. The significant reduction in the average root-mean-square deviation, root-mean-square fluctuation, and effective radius of the DNT with an increase in the NaCl concentration quantifies our observation. We explain how the DNT–ion interactions play a vital role in the conformational fluctuation of the DNT. To understand the salt dependence of the mechanical properties of the DNTs, we have calculated the stretch modulus (γ) and persistence length (LP) as a function of salt concentration. The calculated stretch moduli of the DNTs change from 8.3 to 13 nN, and the persistence length of the DNT varies from 6 to 10 μm when the NaCl salt concentration is varied from 0 to 1 M. Both the stretch modulus and the persistence length calculations reaffirm the structural stability of the DNT at higher salt concentrations. We find similar trends for another monovalent salt (KCl). However, for a divalent salt (MgCl2), we find minimal variation in the structural properties with an increase in the salt concentration.
- RSC
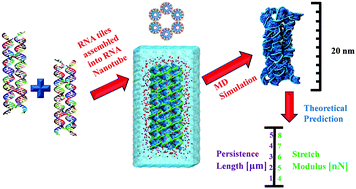 Atomic structures of RNA nanotubes and their comparison with DNA nanotubesSupriyo Naskar, Himanshu Joshi, Banani Chakraborty, and 2 more authorsNanoscale, Jul 2019
Atomic structures of RNA nanotubes and their comparison with DNA nanotubesSupriyo Naskar, Himanshu Joshi, Banani Chakraborty, and 2 more authorsNanoscale, Jul 2019We present a computational framework to model RNA based nanostructures and study their microscopic structures. We model hexagonal nanotubes made of 6 dsRNA (RNTs) connected by double crossover (DX) at different positions. Using several hundred nano-second (ns) long all-atom molecular dynamics simulations, we study the atomic structure, conformational change and elastic properties of RNTs in the presence of explicit water and ions. Based on several structural quantities such as root mean square deviation (RMSD) and root mean square fluctuation (RMSF), we find that the RNTs are almost as stable as DNA nanotubes (DNTs). Although the central portion of the RNTs maintain its cylindrical shape, both the terminal regions open up to give rise to a gating like behavior which can play a crucial role in drug delivery. From the bending angle distribution, we observe that the RNTs are more flexible than DNTs. The calculated persistence length of the RNTs is in the micron range which is an order of magnitude higher than that of a single dsRNA. The stretch modulus of the RNTs from the contour length distribution is in the range of 4–7 nN depending on the sequence. The calculated persistence length and stretch modulus are in the same range of values as in the case of DNTs. To understand the structural properties of RNTs at the individual base-pair level we have also calculated all the helicoidal parameters and analyzed the relative flexibility and rigidity of RNTs having a different sequence. These findings emphasized the fascinating properties of RNTs which will expedite further theoretical and experimental studies in this field.
- APS
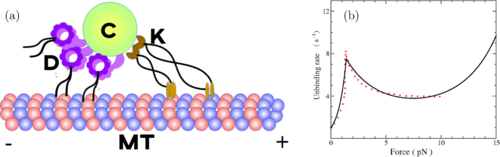 Dynein catch bond as a mediator of codependent bidirectional cellular transportPalka Puri, Nisha Gupta, Sameep Chandel, and 5 more authorsPhysical Review Research, Sep 2019
Dynein catch bond as a mediator of codependent bidirectional cellular transportPalka Puri, Nisha Gupta, Sameep Chandel, and 5 more authorsPhysical Review Research, Sep 2019Intracellular bidirectional transport of cargo on microtubule filaments is achieved by the collective action of oppositely directed dynein and kinesin motors. Experiments have found that in certain cases, inhibiting the activity of one type of motor results in an overall decline in the motility of the cellular cargo in both directions. This counterintuitive observation, referred to as the paradox of codependence, is inconsistent with the existing paradigm of a mechanistic tug of war between oppositely directed motors. Unlike kinesin motors, dynein motors exhibit catch bonding, wherein the unbinding rates of these motors decrease with increasing force on them. Incorporating this catch-bonding behavior of dyneins in a theoretical model, we show that the functional divergence of the two motor species manifests itself as an internal regulatory mechanism, and leads to codependent-transport behavior in biologically relevant regimes. Using analytical methods and stochastic simulations, we analyze the processivity characteristics and probability distribution of run times and pause times of transported cellular cargoes. We show that catch bonding can drastically alter the transport characteristics and also provide a plausible resolution of the paradox of codependence.


